
Background
Le corna di cervo possono rappresentare il 28% del peso corporeo scheletrico[1, 2] e sono l’unico esempio conosciuto di tessuto di mammifero che si rigenera rapidamente, producendo facilmente 10 kg o più di tessuto osseo in un periodo relativamente breve di 2 o 3 mesi [1- 7]. Sebbene le corna di cervo e l’osso umano si sviluppino attraverso modalità di ossificazione intramembrana ed endocondrale[3, 4, 6, 7], le corna di cervo possono crescere fino a 2 cm al giorno [3], in netto contrasto con l’osso del femore umano, che cresce a 2 cm all’anno durante la pubertà [8]. Quindi, se i componenti molecolari che sono coinvolti in questo processo possono essere chiariti, ci si aspetta che questa conoscenza faccia progredire la nostra comprensione della rigenerazione ossea dei mammiferi e promette di generare rapidamente grandi volumi ossei per l’ingegneria del tessuto scheletrico.
Nonostante gli sforzi precedenti[3, 4, 6, 7, 9-20], i geni coinvolti nella rigenerazione rapida delle corna rimangono poco studiati, e manca la dimostrazione funzionale del loro ruolo (o dei loro ruoli) nella proliferazione e nella differenziazione ossea. Ad esempio, studi trascrittomici precedenti di tessuti di corno di cervo utilizzati microarray di topo[12], cDNA-amplificato frammento polimorfismo lunghezza del frammento (cDNA-AFLP)[15], o RNA-seq[18-20], ma gli sforzi per identificare e caratterizzare i contributi genici alla rapida crescita del corno di cervo sono stati ostacolati da problemi logistici e tecnici. Tali limitazioni includono l’ibridazione tra specie incrociate[12], la variazione di sequenza[21], il numero schiacciante di candidati in set di dati trascriptomici[12, 18-20], e la presenza di complesse variabili spaziali e temporali tra i campioni di tessuto [12,15, 18-20]. In effetti, una delle sfide principali quando si lavora con i set di dati trascrittomici è determinare quali geni rilevati svolgono un ruolo nella proliferazione, nella differenziazione scheletrica o in un processo cellulare completamente estraneo. Pertanto, i precedenti studi trascrittomici finora si sono limitati alla profilazione dell’espressione genica con una caratterizzazione poco o nulla del contributo genico alla proliferazione o alla differenziazione dei corni.
In questo studio, abbiamo ipotizzato che un confronto in vitro di daini (FD) e dati RNA-seq umani potrebbe aggirare diverse delle sfide sopra menzionate nell’identificazione dei geni di proliferazione e mineralizzazione dei corni FD. Questo si basava sul nostro ragionamento che il ritmo veloce della rigenerazione del corno di cervo deve derivare, almeno in parte, dalla rapida proliferazione e differenziazione dei progenitori scheletrici. Così, un confronto dei geni espressi in modo differenziato ottenuti da cellule derivate da FD o umane coltivate sotto identiche condizioni di controllo e di gruppo di trattamento eliminerebbe complesse variabili spaziali e temporali in vivo, semplificando al tempo stesso l’analisi bioinformatica per identificare i candidati genici di proliferazione e mineralizzazione espressi in modo univoco nei cervi (file aggiuntivo 1: FiguraS1; file aggiuntivo 2). Le cellule staminali mesenchimali umane (hMSCs) sono state selezionate per essere confrontate con un target terapeutico clinicamente promettente per la medicina rigenerativa a base cellulare[22]. Nei cervi, le corna si rigenerano dai progenitori scheletrici che si trovano nel periostio delle appendici craniche chiamate pediccoli (pedicle periosteo; PP), che, a loro volta, generano cellule mesenchimatiche di riserva (RM) della punta del corno in crescita che alla fine si differenziano in condrociti e cellule ossee mineralizzanti (file aggiuntivo 1: figuraS2) [3, 4, 6, 7]. Sulla base di questo, i progenitori scheletrici sono stati raccolti da daini (FD) periostio facciale (FP; come un non-antler-derivato di controllo), PP, e tessuti RM per determinare un tipo di cellula adatto per il confronto con hMSCs. Successivamente, RNA-seq è stata eseguita, e geni la cui espressione erano unici per i progenitori di corno FD sono stati identificati sulla base di analisi di sottrazione tra FD e set di dati umani. Infine, la funzione e la rilevanza fisiologica di questi geni identificati sono stati determinati da immunofluorescenza, sovraespressione genica e studi di knockdown genico.
Risultati
Caratterizzazione di cellule derivate da corna di cervo
In questo studio, gli studi di caratterizzazione cellulare sono stati eseguiti su progenitori scheletrici di derivazione umana e FD- e per stabilire un modello in vitro per la comparazione RNA-seq. Tra le cellule raccolte, le cellule RM hanno mostrato la più alta attività della fosfatasi alcalina (ALP) in presenza di proteina morfogenetica ossea-2 che induce l’osteogenesi (BMP-2; Fig. 1a) mentre sorprendentemente, le cellule FP e PP coltivate in condizioni di mineralizzazione con BMP-2 e desametasone per 24 giorni hanno mostrato una colorazione rosso alizarina S poco o niente positiva per i depositi di calcio (Fig. 1b). Inoltre, l’analisi FACS (fluorescence-activated cell sorting) e la colorazione in immunofluorescenza hanno indicato che le cellule RM in vitro coltivate erano simili alle loro controparti in vivo[16, 17], con una maggioranza (99,3%) di cellule RM che esprimono ALP e un piccolo sottoinsieme (5,2%) che esprime STRO-1 (Fig. 1c). Insieme, questi studi hanno indicato che in vitro-culturati cellule RM erano modelli ideali di progenitori scheletrici corno di cervo progenitori scheletrici per il confronto con hMSCs.Fig. 1Characterization in vitro-culturati FD-derivato cellule. un FP e le cellule RM (isolare 2) coltivate con 100 ng / ml BMP-2 per 6 giorni esposti maggiore attività ALP rispetto al loro rispettivo controllo, mentre le cellule PP (isolare 2) non ha fatto. Semi-quantificazione dell’attività ALP in FP, PP, e cellule RM. b FP e cellule PP (isolato 2) coltivate con 100 ng / ml BMP-2 e 100 nM desametasone desametasone per 24 giorni non ha mostrato un aumento della colorazione Alizarin Red S rispetto al loro rispettivo controllo. Quantificazione della colorazione Alizarin Red S nelle cellule FP e PP. c Analisi FACS delle cellule RM. La percentuale di cellule che erano negativi per STRO1 e ALP, negativi per STRO1 ma positivi per ALP, negativi per ALP ma positivi per STRO1, e positivi sia per STRO1 e ALP erano 0,28-1,25%, 91,83-97,53%, 0,004-0,10%, e 1,20-7,89%, rispettivamente. STRO1 e colorazione di immunofluorescenza ALP nelle cellule RM. Verde, cellule STRO1-positive. Rosso, cellule ALP-positive. Barre di scala come indicato. I dati provengono da n = 3 isolati (tre esperimenti indipendenti con nove repliche per isolato per gli studi ALP e di mineralizzazione e un esperimento indipendente con tre repliche per isolato per gli studi FACs). I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano l’errore standard della media o SEM. Significato statistico come indicato
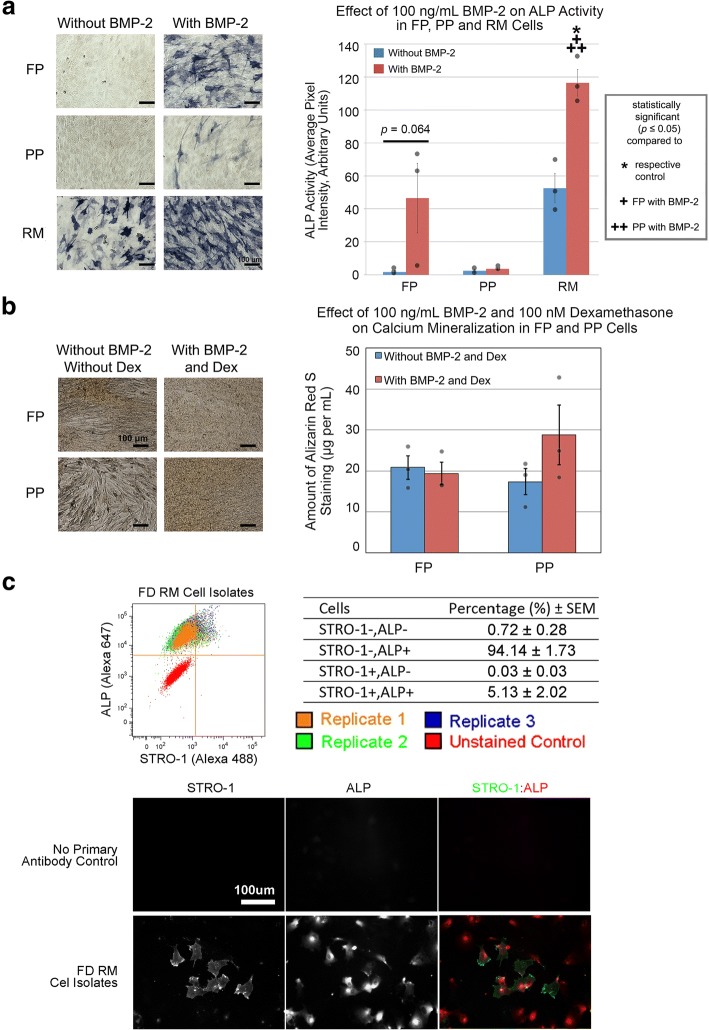
Fig. 1.Caratterizzazione di cellule derivate da FD in vitro. un FP e cellule RM (isolare 2) coltivate con 100 ng / ml BMP-2 per 6 giorni ha mostrato un aumento dell’attività ALP rispetto al loro rispettivo controllo, mentre le cellule PP (isolare 2) non ha fatto. Semi-quantificazione dell’attività ALP in FP, PP, e cellule RM. b FP e cellule PP (isolato 2) coltivate con 100 ng / ml BMP-2 e 100 nM desametasone desametasone per 24 giorni non ha mostrato un aumento della colorazione Alizarin Red S rispetto al loro rispettivo controllo. Quantificazione della colorazione Alizarin Red S nelle cellule FP e PP. c Analisi FACS delle cellule RM. La percentuale di cellule che erano negativi per STRO1 e ALP, negativi per STRO1 ma positivi per ALP, negativi per ALP ma positivi per STRO1, e positivi sia per STRO1 e ALP erano 0,28-1,25%, 91,83-97,53%, 0,004-0,10%, e 1,20-7,89%, rispettivamente. STRO1 e colorazione di immunofluorescenza ALP nelle cellule RM. Verde, cellule STRO1-positive. Rosso, cellule ALP-positive. Barre di scala come indicato. I dati provengono da n = 3 isolati (tre esperimenti indipendenti con nove repliche per isolato per gli studi ALP e di mineralizzazione e un esperimento indipendente con tre repliche per isolato per gli studi FACs). I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano l’errore standard della media o SEM. Significato statistico come indicato
Istituzione di un modello in vitro che mette a confronto le cellule RM derivate dal corno di cervo e le cellule RM derivate dal corno di cervo e gli hMSC
Prima di eseguire l’RNA-seq, è stato necessario caratterizzare in vitro le cellule RM coltivate e gli hMSC per determinare se c’è una forte osteogenesi differenziale in vitro che può riflettere la significativa differenza di crescita ossea in vivo. Dimostrazione di successo di questo fenomeno in vitro giustificherebbe quindi il suo utilizzo per la comparazione RNA-seq. Studi di proliferazione che coinvolgono cellule cresciute in tre diversi mezzi di coltura di mammiferi per 6 giorni hanno dimostrato che le cellule RM hanno mostrato un aumento della crescita rispetto agli hMSC. Le cellule RM hanno prodotto 10,7-45,3 × 104 cellule con un tempo di raddoppio di 17,9-24,7 h mentre hMSCs ha prodotto 1,2-3.9×104 cellule con un tempo di raddoppio di 37,8-60,1 h (Fig. 2a; Bartmann et al. [23] e Schallmoser et al. [24]). Come tale, le cellule RM ha prodotto 8,6-11,7 volte l’aumento del numero di cellule e 2,1-2,4 volte meno tempo di raddoppio rispetto al hMSCs quando controllato per la formulazione media (Fig. 2a). L’analisi del ciclo cellulare ha mostrato risultati simili con una maggiore percentuale di cellule RM in fase di divisione cellulare rispetto ai hMSC (Fig. 2b). Studi di differenziazione hanno dimostrato che le cellule RM non sono state sottoposte a differenziazione adipogenica, ma sono state sottoposte a condrogenesi e osteogenesi (Fig. 2c-e). Negli studi di espressione genica osteogenica, le cellule RM (isolato 2) coltivate in presenza di BMP-2 per 6 giorni hanno mostrato un’elevata upregulation di geni osteogenici tipici come la fosfatasi alcalina ( alp ), l’osteocalcina (ocn ), il fattore-1 specifico per l’osteoblasto ( osf-1 )e il fattore di trascrizione 2 (runx2) relativo al controllo di 651,1 volte, 1,9 volte, 42,8 volte e 557,5 volte, rispettivamente (Fig. 2d). Negli studi di mineralizzazione osteogenica, gruppi di controllo per le cellule RM e hMSC (coltivate in assenza di BMP-2 e desametasone) ha esposto 28,0 μg/mL e 39.6 μg/mL Alizarin Red S stain, rispettivamente, mentre i gruppi di trattamento per le cellule RM e hMSC (coltivate in presenza di BMP-2 e desametasone) hanno mostrato 1702,2 μg/mL e 98,0 μg/mL Alizarin Red S stain, rispettivamente (Fig. 2e). Notevolmente, anche se sono state utilizzate condizioni di coltura identiche, le cellule RM e hMSC hanno dimostrato un aumento di 62,3 volte e 1,9 volte, rispettivamente, nella mineralizzazione del calcio rispetto ai loro rispettivi controlli, con le cellule RM che hanno dimostrato 17,4 volte maggiori livelli di mineralizzazione del calcio rispetto ai hMSC, nonostante i livelli di base simili nei gruppi di controllo (Fig. 2e). Inoltre, le cellule RM hanno mostrato 2,0-18,1 volte l’aumento rispetto a hMSC-mediata mineralizzazione del calcio, nonostante una diminuzione di 5 volte la concentrazione di BMP-2, così come 3,1 volte l’aumento della mineralizzazione del calcio rispetto alle cellule hMSC-come C3H10T1/2, nonostante un 1,6 volte più lungo tempo di raddoppio delle cellule (Fig. 2; file aggiuntivo 1: Figura S3; Keret al. [25]). Così, le capacità di proliferazione e mineralizzazione fortemente contrastanti delle cellule RM e hMSCs hanno giustificato l’uso di questo modello in vitro e ci si aspettava che identificassero i geni che orchestrano la crescita differenziale e i tassi di mineralizzazione osservati tra le corna di cervo e l’osso umano.Fig. 2 Le cellule RM mostrano un aumento della proliferazione e della differenziazione osteogenica rispetto alle hMSCs . b L’analisi del ciclo cellulare ha mostrato una maggiore proporzione di cellule RM in fase di divisione cellulare rispetto ai hMSC. c Le cellule RM erano capaci di differenziazione condrogenica ma non adipogenica. d Le cellule RM (isolato 2) coltivate con 100 ng/mL BMP-2 per 6 giorni hanno mostrato un aumento dell’espressione genica osteogenica rispetto al loro rispettivo controllo. e Le cellule RM coltivate con 100 ng/mL BMP-2 e 100 nM desametasone per 24 giorni hanno mostrato un aumento della colorazione Alizarin Red S rispetto ai hMSC. Barre di scala come indicato. I dati provenivano da n = 3 isolati (tre esperimenti indipendenti con nove repliche per isolato per studi di proliferazione e condrogenici, adipogenici e di mineralizzazione e un esperimento indipendente con tre repliche per isolato per studi sul ciclo cellulare) o n = 1 isolato (due esperimenti indipendenti con sei repliche per studi di espressione genica osteogenica). I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano SEM. Significato statistico come indicato
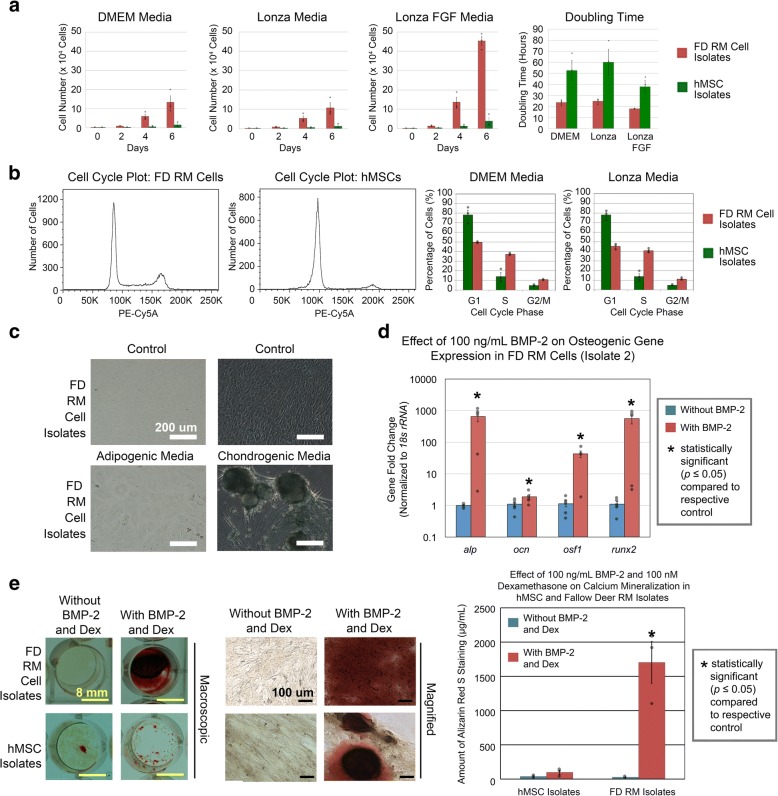
Fig. 2.Le cellule RM mostrano una maggiore proliferazione e differenziazione osteogenica rispetto ai hMSC. a Le cellule RM hanno mostrato una maggiore proliferazione rispetto ai hMSC. b L’analisi del ciclo cellulare ha mostrato una maggiore proporzione di cellule RM in fase di divisione cellulare rispetto ai hMSC. c Le cellule RM erano in grado di differenziazione condrogenica ma non adipogenica. d Le cellule RM (isolato 2) coltivate con 100 ng/mL BMP-2 per 6 giorni hanno mostrato un aumento dell’espressione genica osteogenica rispetto al loro rispettivo controllo. e Le cellule RM coltivate con 100 ng/mL BMP-2 e 100 nM desametasone per 24 giorni hanno mostrato un aumento della colorazione Alizarin Red S rispetto ai hMSC. Barre di scala come indicato. I dati provenivano da n = 3 isolati (tre esperimenti indipendenti con nove repliche per isolato per studi di proliferazione e condrogenici, adipogenici e di mineralizzazione e un esperimento indipendente con tre repliche per isolato per studi sul ciclo cellulare) o n = 1 isolato (due esperimenti indipendenti con sei repliche per studi di espressione genica osteogenica). I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano SEM. Significato statistico come indicato
Identificazione dei geni di proliferazione e mineralizzazione del corno di cervo utilizzando RNA-seq comparativo
Per identificare i geni di proliferazione e mineralizzazione che contribuiscono alla rapida crescita e differenziazione osservata nel nostro modello in vitro, abbiamo focalizzato la nostra attenzione sui geni espressi in modo unico nelle cellule RM. Questo è stato ottenuto confrontando in modo indipendente i dati RNA-seq delle cellule RM (isolato 2) e hMSCs (isolato 24268) in condizioni di proliferazione (gruppo di controllo, 0% siero; gruppo di trattamento, 10% siero) così come la mineralizzazione (gruppo di controllo, 0 ng/mL BMP-2 e 0 nM desametasone; gruppo di trattamento, 100 ng/mL BMP-2 e 100 nM desametasone) (Figg. 3 e 4). I campioni di RNA-seq proliferazione sono stati sequenziati a 76.781.962-99.716.096 letture per biblioteca con repliche che mostrano una forte correlazione dell’espressione genica in condizioni di assenza di siero e contenenti siero (Fig. 3a e file aggiuntivo 1: Tabella S1). Come ci si aspettava da un organismo non modello, una percentuale maggiore di geni non preannunciati era presente in FD (36%) rispetto ai dati RNA-seq umani (8%) (Fig. 3a). Nonostante questo, l’Ingenuity Pathway Analysis (IPA) di trascrizioni annotate ha mostrato un’attivazione simile di percorsi associati alla proliferazione, come i ruoli mitotici di polo-like kinase così come l’espressione di geni tipici della proliferazione come cdk1, rrm1, cdc7, aurka, e plk4 in entrambi i dataset (Fig. 3a). Corrispondentemente, l’analisi dell’ontologia genica ha mostrato l’upregulation dei processi associati alla proliferazione, compresi i punti di controllo mitotici e la condensazione dei cromosomi (Fig. 3b). Inoltre, i campioni di mineralizzazione RNA-seq sono stati sequenziati a 62.601.720-86.750.048 letture per biblioteca con repliche che mostrano una forte correlazione di espressione genica in condizioni di non mineralizzazione e mineralizzazione (Fig. 4a e file aggiuntivo 1: Tabella S2). Analogamente al set di dati sulla proliferazione, una percentuale maggiore di geni non preannunciati era presente in FD (41%) rispetto ai dati RNA-seq umani (13%) (Fig. 4a). IPA di trascrizioni annotate ha mostrato un’attivazione simile di percorsi associati all’osteogenicità, come il ruolo degli osteoblasti, osteoclasti e condrociti nell’artrite reumatoide, così come l’espressione di geni osteogenici tipici come dlx5, tsc22d3, alpl, klf4, ext1, e stc1 in entrambi i dataset (Fig. 4a). Corrispondentemente, l’analisi dell’ontologia genetica ha mostrato upregulation dei processi associati con il catabolismo scheletrico tra cui la sintesi del collagene così come la morfogenesi del viso e del corpo (Fig. 4b). Successivamente, l’analisi di sottrazione è stata eseguita tra i set di dati umani e FD per i geni espressi in modo differenziato. Utilizzando i seguenti criteri di geni FD altamente upregolati (> 5 volte) ed espressi in modo univoco, sono stati identificati 40 geni candidati alla proliferazione e 91 geni candidati alla mineralizzazione (Figg. 3a e 4a). Così, in vitro comparativo RNA-seq identificato candidati genetici che sono stati espressi in modo univoco nelle cellule RM con un ruolo presunto nella rigenerazione rapida corno di cervo. Fig. 3RNA-seq analisi di cellule RM e hMSCs in condizioni di proliferazione e mineralizzazione. un RNA-seq analisi di cellule RM (isolato 2) e hMSCs (isolato 24268) in condizioni di assenza di siero (0% di siero) e contenente siero (10% di siero) ha identificato 40 geni candidati di proliferazione. Gli scatterplot indicano la correlazione(r2) tra i replicati per ogni condizione. FPKM, frammenti per kilobase di trascrizione per milione di letture mappate. b Analisi dell’arricchimento ontologico dei geni delle cellule RM (isolato 2) e hMSC (isolato 24268) in condizioni di proliferazione. I grafici indicano i primi 5 upregulated top 5 (rosso) e downregulated (verde) processi biologici, componenti cellulari, e le funzioni molecolari. I dati sono stati da n = 1 isolato (un esperimento indipendente con due repliche per gruppo) Fig. 4RNA-seq analisi delle cellule RM e hMSCs in condizioni di proliferazione e mineralizzazione. un’ analisi RNA-seq di cellule RM (isolato 2) e hMSC (isolato 24268) sotto controllo (0 ng/mL BMP-2 e 0 nM desametasone) e osteogenico (100 ng/mL BMP-2 e 100 nM desametasone) condizioni identificato 91 geni di mineralizzazione candidati. Gli scatterplot indicano la correlazione(r2) tra i replicati per ciascuna condizione. FPKM, frammenti per kilobase di trascrizione per milione di letture mappate. b Analisi dell’arricchimento ontologico dei geni delle cellule RM (isolato 2) e hMSCs (isolato 24268) in condizioni di mineralizzazione. I grafici indicano i primi 5 upregulated (rosso) e downregulated (verde) processi biologici, componenti cellulari e funzioni molecolari. I dati sono stati da n = 1 isolato (un esperimento indipendente con due repliche per gruppo)
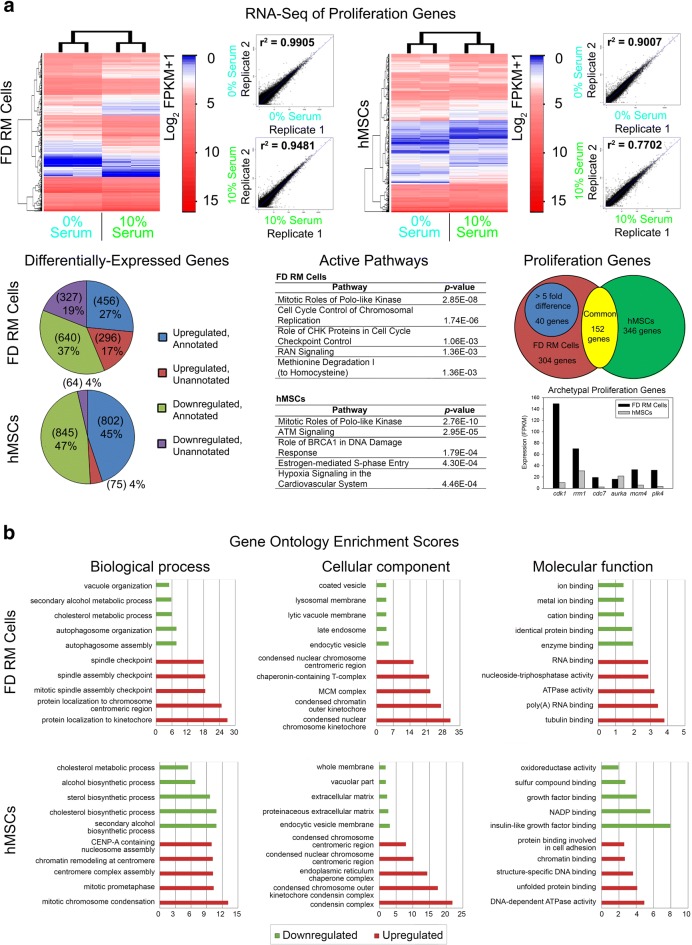
Fig. 3.Fig. 3. Analisi RNA-seq delle cellule RM e hMSC in condizioni di proliferazione e mineralizzazione. un’ analisi RNA-seq delle cellule RM (isolato 2) e hMSC (isolato 24268) in condizioni di assenza di siero (0% di siero) e contenenti siero (10% di siero) ha identificato 40 geni candidati alla proliferazione. Gli scatterplot indicano la correlazione(r2) tra i replicati per ogni condizione. FPKM, frammenti per kilobase di trascrizione per milione di letture mappate. b Analisi dell’arricchimento ontologico dei geni delle cellule RM (isolato 2) e hMSC (isolato 24268) in condizioni di proliferazione. I grafici indicano i primi 5 upregulated top 5 (rosso) e downregulated (verde) processi biologici, componenti cellulari, e le funzioni molecolari. I dati sono stati da n = 1 isolato (un esperimento indipendente con due repliche per gruppo)
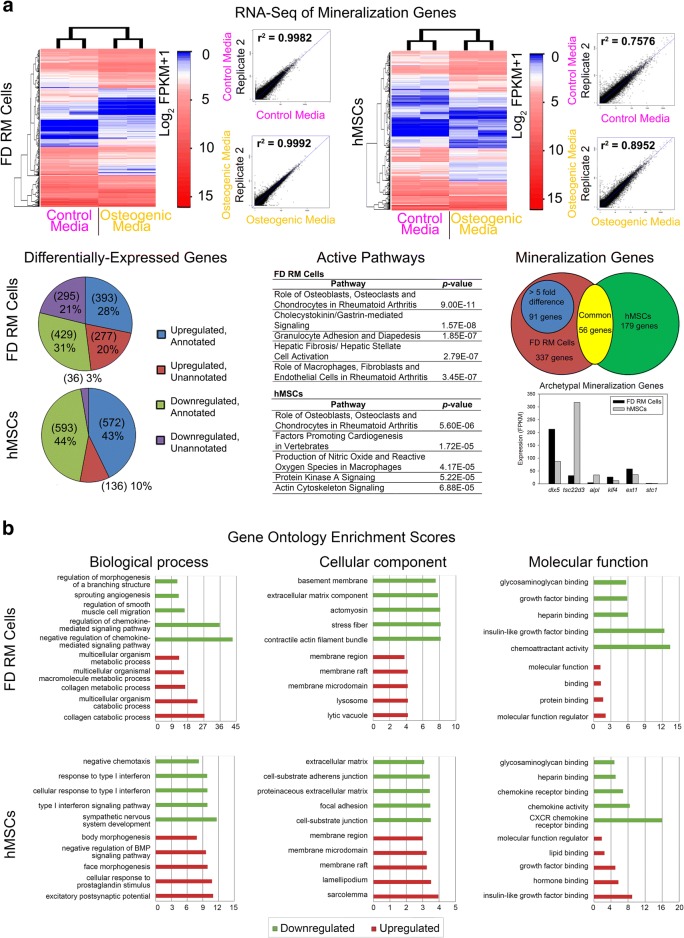
Fig. 4.Fig. 4. Analisi RNA-seq delle cellule RM e hMSC in condizioni di proliferazione e mineralizzazione. un’ analisi RNA-seq delle cellule RM (isolato 2) e hMSC (isolato 24268) sotto controllo (0 ng/mL BMP-2 e 0 nM desametasone) e osteogeniche (100 ng/mL BMP-2 e 100 nM desametasone) ha identificato 91 geni di mineralizzazione candidati. Gli scatterplot indicano la correlazione(r2) tra i replicati per ciascuna condizione. FPKM, frammenti per kilobase di trascrizione per milione di letture mappate. b Analisi dell’arricchimento ontologico dei geni delle cellule RM (isolato 2) e hMSCs (isolato 24268) in condizioni di mineralizzazione. I grafici indicano i primi 5 upregulated (rosso) e downregulated (verde) processi biologici, componenti cellulari e funzioni molecolari. I dati sono stati da n = 1 isolato (un esperimento indipendente con due repliche per gruppo)
Convalida di RNA-seq comparativo in vitro
Per convalidare la rilevanza fisiologica e il ruolo dei geni espressi in modo univoco identificati da RNA-seq comparativo in vitro, è stato selezionato un candidato genico di proliferazione e mineralizzazione, che è stato ulteriormente valutato in immunofluorescenza, sovraespressione genica e studi di knockdown genico (Figg. 5 e 6). Dal momento che le trasfezioni di hMSCs non hanno avuto successo (dati non mostrati), le cellule di topo hMSC-come il topo C3H10T1/2 sono state utilizzate in studi di sovraespressione genica.Fig. 5Identificazione di uhrf1 come un gene di proliferazione espresso in modo univoco utilizzando RNA-seq comparativo in vitro. a colorazione di immunofluorescenza UHRF1 in tessuto rigenerante di corno di cervo. b Le cellule RM coltivate con 30 nM uhrf1 siRNA per 3 giorni hanno mostrato una diminuzione della proliferazione rispetto al controllo mock-transfected. c Le cellule C3H10T1/2 stabilmente trasfettate con uhrf1 hanno mostrato un aumento della proliferazione rispetto al controllo non trasformato e al controllo del plasmide vuoto. C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con uhrf1 mantenuto l’inibizione di contatto. Curve di crescita rappresentativi sono mostrati. d C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettato con uhrf1 e coltivato con 100 ng / ml BMP-2 per 6 giorni ha mostrato un aumento dell’attività ALP rispetto al controllo non trasformato e il controllo del plasmide vuoto. Barre di scala come indicato. I dati provenivano da n==3 isolati (un branco indipendente per studi di immunofluorescenza delle corna) o n==3 esperimenti indipendenti con nove repliche per gruppo per gli studi sulla proliferazione di uhrf1 knockdown e sovraespressione e sulla differenziazione degli osteoblasti. I cerchi grigi indicano i punti di dati osservati. Barre di errore indicano SEM. Significato statistico come indicatoFig. 6Identificazione 6Identificazione di s100a10 come un gene di mineralizzazione espresso in modo univoco utilizzando in vitro comparativo RNA-seq. un S100A10 immunofluorescenza colorazione S100A10 in tessuto rigenerante corno di cervo. b Le cellule RM (isolate 2) coltivate con 100 ng/mL BMP-2 e 100 nM desametasone hanno mostrato un aumento dell’espressione di S100A10 rispetto al controllo. c Le cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 4 h hanno mostrato un aumento dell’espressione del gene alp rispetto al controllo non trasformato e al controllo del plasmide vuoto. C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 12 giorni hanno mostrato un aumento dell’espressione del gene ocn e runx2 rispetto al loro rispettivo controllo. d C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 4 giorni hanno mostrato un aumento dell’attività ALP rispetto al controllo non trasformato. e C3H10T1/2 cellule stabilmente trasfettate con s100a10 e coltivate in presenza di 100 ng/mL BMP-2 e 100 nM desametasone esposto un aumento della colorazione Alizarin Red S rispetto al controllo non trasformato e al controllo del plasmide vuoto. Barre di scala come indicato. I dati sono stati ottenuti da n = 3 isolati (un branco indipendente per studi di immunofluorescenza del corno), n = 2-3 esperimenti indipendenti con 4-10 repliche per gruppo per studi di espressione genica osteogenica, n = 2-3 esperimenti indipendenti con 4-10 repliche per gruppo per studi di espressione genica osteogenica, n = 3 isolati (un branco indipendente per studi di immunofluorescenza del corno).= 3 esperimenti indipendenti con 9 repliche per gruppo per studi di ALP di sovraespressione s100a10, e n = 5 esperimenti indipendenti con 15 repliche per gruppo per studi di mineralizzazione di sovraespressione s100a10. I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano SEM. Significato statistico come indicato
Dei 40 candidati al gene della proliferazione, FD uhrf1 è stato scelto per il suo ruolo nell’ereditarietà epigenetica[26] e l’alta espressione in diversi tumori[27], suggerendo un ruolo per questo gene nel controllo simultaneo dell’auto-rinnovamento delle cellule staminali[28] e della crescita nelle corna dei cervi. Negli studi di immunofluorescenza, le corna rigeneranti FD ottenute da un gregge indipendente hanno mostrato un’espressione di UHRF1 nel tessuto RM (Fig. 5a), mentre l’integrazione di mitogeni noti come il fattore di crescita dei fibroblasti-2 (FGF-2) e il fattore di crescita insulino-simile-1 (IGF-1) nelle cellule RM ha mostrato una buona correlazione tra l’espressione di UHRF1 e la proliferazione delle cellule RM (file aggiuntivo 1: Figura S4). Ad esempio, le cellule RM hanno mostrato un aumento dell’espressione UHRF1 rispetto al controllo quando sono state coltivate in presenza di IGF-1 in condizioni di assenza di siero e in presenza di FGF-2 sia in condizioni di assenza di siero che in condizioni di presenza di siero (file aggiuntivo 1: FiguraS4a e S4b). Corrispondentemente, l’aumento della proliferazione è stato osservato solo nelle condizioni di coltura sopra menzionate, ma non in presenza di IGF-1 in condizioni sieriche, dove non c’era upregulation di UHRF1 (Additional file 1: Figura S4a e S4bsp; 1: FiguraS4c). Insieme, questi dati dimostrano la rilevanza fisiologica di uhrf1 nella proliferazione cellulare FD RM e verificato il modello di espressione di questo gene rispetto ai nostri dati di RNA-seq proliferazione (Fig. 5ae file aggiuntivo 1: Figura S4). Negli studi sul gene knockdown, le cellule RM (isolato 2) trattate con siRNA di controllo hanno prodotto 5,1×104 cellule,mentre i gruppi trattati con siRNA hanno prodotto 2,8-4,2×104 cellule (Fig. 5b e Additional file 1: Figura S5a). Come tale, siRNA-mediato knockdown siRNA-mediata di uhrf1 inibito la crescita delle cellule RM del 17,9-45,5% (Fig. 5b e file aggiuntivo 1: Figura S5a). Negli studi di sovraespressione genica, le cellule C3H10T1/2 e C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con plasmide vuoto ha prodotto 7,7-9,1 × 104 cellule con un tempo di raddoppio di 13.9-14,3 h mentre le cellule C3H10T1/2 stabilmente trasfettate con uhrf1 hanno prodotto 13,6×104 cellule con un tempo di raddoppio di 11,8 h (Fig. 5c). Come tale, la sovraespressione di FD uhrf1 in C3H10T1/2 cellule (file aggiuntivo 1: FiguraS5b) ha aumentato la proliferazione cellulare di 1,49-1,76 volte e diminuito il tempo di raddoppio delle cellule da 13.8-15,6% (Fig. 5c)senza compromettere l’inibizione del contatto (Fig. 5c e file aggiuntivo 1: Figura S6) o attività ALP (Fig. 5e). Questo è particolarmente degno di nota, dato che le cellule C3H10T1/2 non trasformate crescono già rapidamente e raddoppiano ogni 13,9-15,1 ore (Fig. 5c e Ker etal. [25]). Così, in vitro comparativo RNA-seq identificato uhrf1 come contributo ad un ruolo precedentemente sconosciuto nella proliferazione delle cellule corno FD.
Dei 91 candidati al gene della mineralizzazione, FD s100a10, che ha ruoli nella fibrinolisi e nell’organizzazione della membrana intracellulare[29], è stato scelto a causa della scarsità di dati relativi al suo ruolo nell’osteogenesi, che lo rende un nuovo obiettivo per ulteriori studi. Inoltre, i membri della famiglia delle proteine S100 come S100A4 sono stati segnalati come un regolatore negativo della differenziazione degli osteoblasti e della mineralizzazione della matrice[30]. Negli studi di immunofluorescenza, le corna FD rigeneranti ottenute da un allevamento indipendente hanno mostrato l’espressione di S100A10 nelle regioni cartilaginee del corno in fase di mineralizzazione (Fig. 6a) mentre S100A10 è stato upregulated durante la differenziazione osteogenica delle cellule RM (Fig. 6b). Insieme, questi dati dimostrano la rilevanza fisiologica di s100a10 nella mineralizzazione FD e hanno verificato il modello di espressione di questo gene rispetto ai nostri dati di mineralizzazione RNA-seq (Fig. 6a, b). Negli studi di sovraespressione genica, C3H10T1/2 stabilmente trasfettato con s100a10 (file aggiuntivo 1: FiguraS7) ha mostrato un aumento dell’espressione alpina (Fig. 6c) e dell’attività ALP (Fig. 6d) rispetto al plasmidevuoto e ai controlli non trasformati, così come l’upregulation di altri geni osteogenici tipici ocn e runx2 rispetto al controllo quando coltivati in presenza di BMP-2 (Fig. 6c). Inoltre, C3H10T1/2 stabilmente trasfettato con s100a10 ha mostrato un aumento della mineralizzazione del calcio rispetto ai controlli plasmidici non trasformati e vuoti quando in presenza di BMP-2 e desametasone (Fig. 6e). In queste condizioni di coltura, le cellule C3H10T1/2, C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con plasmide vuoto, e C3H10T1/2 cellule stabilmente trasfettate con s100a10 esposto 257,2 μg/mL, 341,9 μg/mL, e 807,9 μg/mL Alizarin Red S macchia, rispettivamente (Fig. 6e). Come tale, la sovraespressione di FD s100a10 in cellule C3H10T1/2 ha aumentato la mineralizzazione mediata dalle cellule di 2,4-3,1 volte (Fig. 6e). Così, in vitro RNA comparativo RNA-seq identificato s100a10 come contributo verso un ruolo precedentemente sconosciuto nella mineralizzazione cellulare FD corno corno. Insieme, questi risultati hanno dimostrato la capacità dell’analisi comparativa in vitro RNA-seq per identificare i geni di proliferazione e mineralizzazione FD espressi in modo univoco.
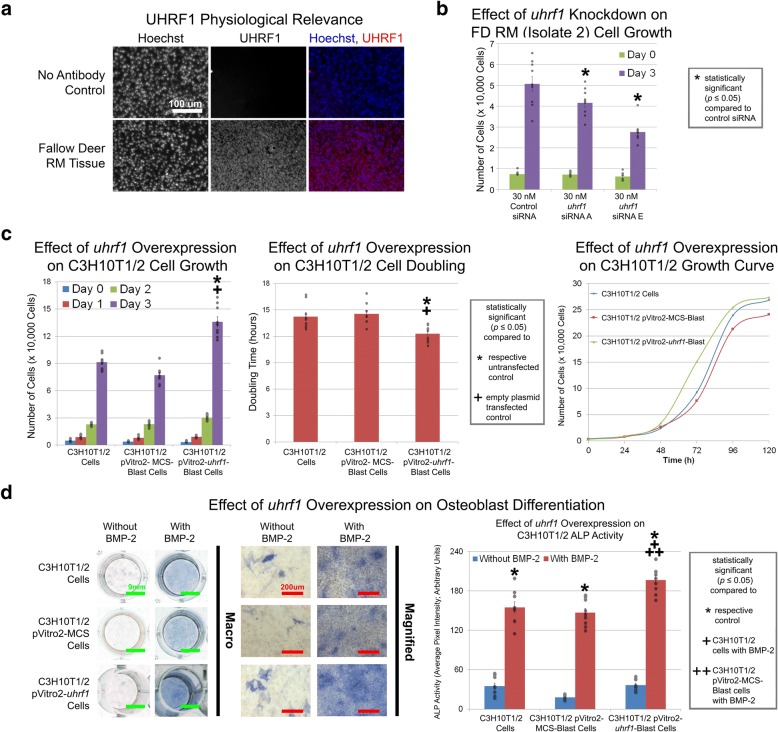
Fig. 5.Fig. 5. Identificazione di uhrf1 come un gene di proliferazione espresso in modo univoco utilizzando RNA-seq comparativo in vitro. a UHRF1 immunofluorescenza colorazione UHRF1 nel tessuto rigenerante corno di cervo. b Le cellule RM coltivate con 30 nM uhrf1 siRNA per 3 giorni hanno mostrato una diminuzione della proliferazione rispetto al controllo mock-transfected. c C3H10T1/2 cellule stabilmente trasfettate con uhrf1 hanno mostrato una maggiore proliferazione rispetto al controllo non trasformato e il controllo del plasmide vuoto. C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con uhrf1 mantenuto l’inibizione di contatto. Curve di crescita rappresentativi sono mostrati. d C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettato con uhrf1 e coltivato con 100 ng / ml BMP-2 per 6 giorni ha mostrato un aumento dell’attività ALP rispetto al controllo non trasformato e il controllo del plasmide vuoto. Barre di scala come indicato. I dati provenivano da n==3 isolati (un branco indipendente per studi di immunofluorescenza delle corna) o n==3 esperimenti indipendenti con nove repliche per gruppo per gli studi sulla proliferazione di uhrf1 knockdown e sovraespressione e sulla differenziazione degli osteoblasti. I cerchi grigi indicano i punti di dati osservati. Barre di errore indicano SEM. Significato statistico come indicato
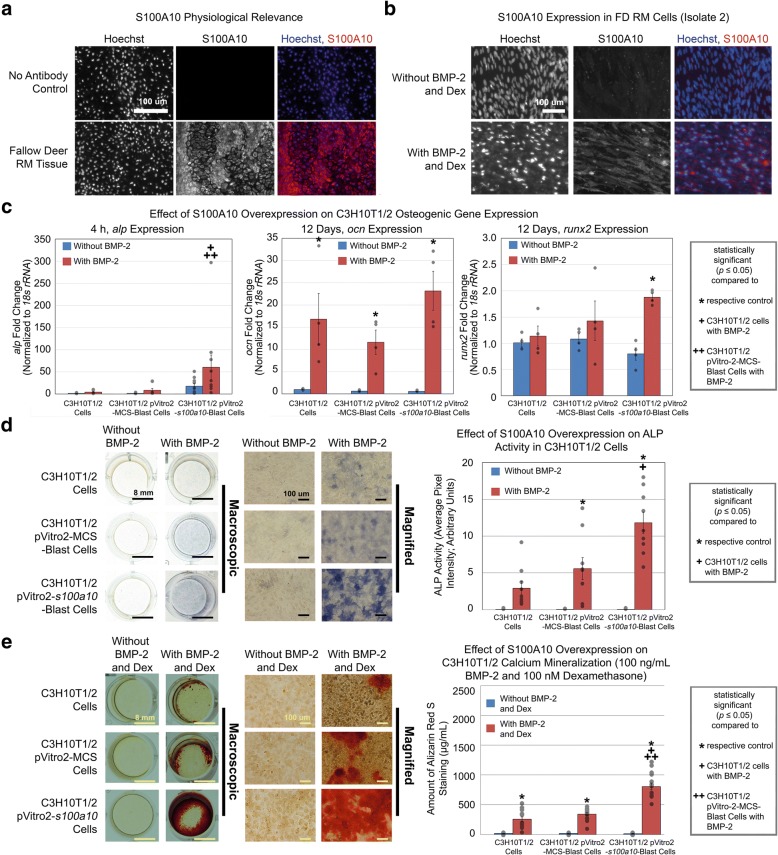
Fig. 6.Fig. 6. Identificazione di s100a10 come un gene di mineralizzazione espresso in modo univoco utilizzando RNA comparativo in vitro-seq. a S100A10 colorazione di immunofluorescenza nel tessuto rigenerante corno di cervo. b Le cellule RM (isolato 2) coltivate con 100 ng/mL BMP-2 e 100 nM desametasone hanno mostrato un aumento dell’espressione di S100A10 rispetto al controllo. c C 3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 4 h hanno mostrato un aumento dell’espressione del gene Alp rispetto al controllo non trasformato e al controllo del plasmide vuoto. C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 12 giorni hanno mostrato un aumento dell’espressione del gene ocn e runx2 rispetto al loro rispettivo controllo. d C3H10T1/2 cellule C3H10T1/2 stabilmente trasfettate con s100a10 e coltivate con 100 ng/mL BMP-2 per 4 giorni hanno mostrato un aumento dell’attività ALP rispetto al controllo non trasformato. e C3H10T1/2 cellule stabilmente trasfettate con s100a10 e coltivate in presenza di 100 ng/mL BMP-2 e 100 nM desametasone esposto un aumento della colorazione Alizarin Red S rispetto al controllo non trasformato e al controllo del plasmide vuoto. Barre di scala come indicato. I dati sono stati ottenuti da n = 3 isolati (un branco indipendente per studi di immunofluorescenza del corno), n = 2-3 esperimenti indipendenti con 4-10 repliche per gruppo per studi di espressione genica osteogenica, n = 2-3 esperimenti indipendenti con 4-10 repliche per gruppo per studi di espressione genica osteogenica, n = 3 isolati (un branco indipendente per studi di immunofluorescenza del corno).= 3 esperimenti indipendenti con 9 repliche per gruppo per studi di ALP di sovraespressione s100a10, e n = 5 esperimenti indipendenti con 15 repliche per gruppo per studi di mineralizzazione di sovraespressione s100a10. I cerchi grigi indicano i punti di dati osservati. Le barre di errore indicano SEM. Significato statistico come indicato
Discussione
In questo studio, le cellule RM derivate dal corno di cervo e le cellule staminali mesenchimali derivate dal midollo osseo umano sono state utilizzate per stabilire un modello in vitro per la comparazione RNA-seq (file aggiuntivo 1: figure S1, S2 e S3; figure.1 e 2) e hanno identificato uhrf1 e s100a10 come proliferazione di corna di cervo espressa in modo univoco (Figg. 3 e 5 e file aggiuntivo 1: Figure S4, S5 e S6) e geni di mineralizzazione (Figg. 4 e 6 e file aggiuntivo 1: Figura S7), rispettivamente. L’approccio qui sviluppato può essere ampiamente applicato allo studio di un altro fenomeno biologico, e i geni identificati con questo approccio non solo farà progredire la nostra comprensione della rigenerazione ossea dei mammiferi, ma offrirà anche promettenti strategie terapeutiche per l’ingegneria del tessuto osseo.
La nostra premessa per l’utilizzo di un approccio in vitro si basava su diversi motivi. In primo luogo, un approccio in vitro ha permesso un maggiore controllo sperimentale, consentendo alle condizioni di coltura di definire e studiare il fenomeno di interesse. In secondo luogo, l’uso di condizioni identiche per la coltura di cervi e cellule umane non solo ha eliminato le complesse variabili spaziali e temporali in vivo, ma ha anche permesso che i dati di espressione genica differenziale di ogni specie servissero come base di confronto per identificare i geni di corno di cervo espressi in modo univoco attraverso una semplice analisi di sottrazione. Ad esempio, sono state utilizzate condizioni di media prive di siero e contenenti siero per identificare i geni di proliferazione attraverso l’espressione genica differenziale. Questi esperimenti sono eseguiti in modo indipendente per le cellule umane e di cervo. In seguito, l’analisi di sottrazione tra i geni della proliferazione umana e dei cervi produrrebbe geni di proliferazione dei cervi espressi in modo univoco. Inoltre, un approccio in vitro ha ridotto l’onere logistico per l’alloggiamento a lungo termine di un grande organismo non modellato come il cervo, mentre l’RNA-seq ha permesso il rilevamento completo e sensibile delle trascrizioni[31] con poca parzialità ed errori anche quando vengono utilizzati genomi di riferimento di specie non bersaglio[32]. Pertanto, ci si aspettava un approccio in vitro per identificare i geni di proliferazione e mineralizzazione dei corni di cervo espressi in modo univoco.
Nello stabilire questo modello per identificare i geni di proliferazione e mineralizzazione del corno di cervo, sono state confrontate cellule hMSC e cellule RM. La base alla base di questa scelta deriva dalle differenze significative nella crescita ossea tra i tessuti scheletrici umani e i tessuti del corno di cervo[3, 4, 6-8]così come la promessa terapeutica di ricapitolare tale rapida crescita nei tessuti scheletrici umani. Anche se gli hMSC e le cellule RM hanno un background genetico diverso e non provengono da tessuti anatomicamente equivalenti (gli hMSC sono stati ottenuti dalla cresta iliaca, mentre le cellule RM sono state raccolte dal peduncolo cranico), un confronto trascrittomico di queste cellule dovrebbe ancora fornire importanti informazioni sui geni necessari per stimolare la proliferazione rapida e l’alta mineralizzazione, soprattutto perché gli hMSC sono un obiettivo terapeutico clinicamente promettente per l’ingegneria del tessuto osseo[22, 33]. Inoltre, le cellule RM coltivate in vitro hanno mostrato un’elevata capacità osteogenica (File aggiuntivo 1: FiguraS3, Figg. 1 e 2) e simili marcatori di superficie cellulare (ALP e STRO-1) come loro controparti in vivo [16, 17]. Rispetto ai hMSC in condizioni di coltura identiche, le cellule RM hanno dimostrato 8,6-11,7 volte maggiore crescita delle cellule e 17,4 volte maggiori livelli di mineralizzazione del calcio (Fig. 2), in una certa misura che riflette la rapida crescita e fenomeni di differenziazione osservati nella rigenerazione delle corna di cervo. Insieme, questi studi hanno giustificato l’uso di hMSCs in vitro coltivate e cellule RM per identificare la proliferazione delle corna di cervo e dei geni di mineralizzazione.
Per identificare i geni di proliferazione e mineralizzazione del corno di cervo, è stato eseguito l’RNA-seq. Il confronto e la successiva analisi di sottrazione dei trascrittomi in condizioni di proliferazione e mineralizzazione hanno identificato 40 geni di proliferazione e 91 geni di mineralizzazione che sono stati espressi in modo univoco nelle cellule RM (Fig. 3 e Fig. 4). Ulteriori analisi bioinformatiche utilizzando database commerciali e pubblici hanno mostrato l’attivazione e l’arricchimento dei percorsi di proliferazione e mineralizzazione o parole chiave, in accordo con le condizioni di coltura utilizzate (Figs. 3 e 4). Per convalidare il contributo dei geni identificati, uhrf1 e s100a10 sono stati scelti per ulteriori studi in base al loro ruolo potenziale nel rinnovamento delle cellule staminali[28] o novità, rispettivamente. Sebbene la loro partecipazione alla biologia del corno di cervo non sia stata riportata, altri studi hanno indicato che UHRF1 è coinvolto nella proliferazione e maturazione delle celluleTreg del colon attraverso il silenziamento epigenetico del CDKNA/P21, un inibitore dei complessi chinasi ciclina/ciclino-dipendente[34] mentre i membri correlati della famiglia delle proteine S100 come S100A4 sono coinvolti nella regolazione negativa della differenziazione degli osteoblasti[30]. La sovraespressione di FD uhrf1 in una linea cellulare del topo con caratteristiche simili all’hMSC[25] ha aumentato la proliferazione cellulare senza influenzare l’inibizione del contatto (Fig. 5, file aggiuntivo 1: figure S5 e S6) mentre il knockdown mediato da siRNA di uhrf1 nelle cellule RM ha diminuito la crescita cellulare (Fig. 5 efile aggiuntivo 1: figura S5). Allo stesso modo, la sovraespressione di FD s100a10 in questa linea cellulare del topo ha aumentato l’espressione genica osteogenica, l’attività ALP, e la mineralizzazione del calcio (Fig. 6 e file aggiuntivo 1: Figura S7). Inoltre, la rilevanza fisiologica di questi risultati è stata confermata dalla colorazione in immunofluorescenza delle corna di cervo rigenerante (Figg. 5 e 6). Così, in vitro comparativo RNA-seq identificato in vitro RNA-seq proliferazione corna di cervo e geni mineralizzazione.
Il successo dell’approccio comparativo RNA-seq in vitro dipende fortemente da diversi fattori. In primo luogo, è necessario utilizzare adeguate condizioni di coltura in vitro che modellano da vicino il fenomeno biologico di interesse. In questo studio, abbiamo dimostrato che le cellule RM proliferavano rapidamente ed esibivano un aumento dei livelli di mineralizzazione del calcio rispetto agli hMSC, che simulavano il fenomeno della rapida crescita ossea nelle corna dei cervi (Fig. 2). Tuttavia, è possibile che le condizioni di coltura non riflettano accuratamente gli stimoli di crescita e di differenziazione nella rigenerazione delle corna di cervo, dando luogo a falsi positivi o negativi. Come tale, è fondamentale accertare la rilevanza fisiologica di questi risultati determinando l’espressione genica o proteica nel tessuto biologico rilevante (Figg. 5 e 6). Nonostante queste limitazioni, il nostro approccio è riuscito a identificare uhrf1 e s100a10 come geni precedentemente sconosciuti FD proliferazione corno e mineralizzazione, rispettivamente. Tuttavia, ulteriori modifiche a questo studio possono estendere ulteriormente il suo impatto. Queste includono condizioni di coltura alternative che imitano meglio le condizioni fisiologiche, come l’uso di co-colture per studiare le interazioni paracrine dell’osso del corno e del velluto (pelle), nonché l’applicazione di criteri di selezione più rigorosi come il confronto di dati RNA-seq in vitro (il nostro studio attuale) e in vivo (tessuto del corno di cervo). In secondo luogo, è importante riconoscere che l’RNA-seq comparativo in vitro esegue l’analisi di sottrazione tra i geni RM espressi in modo differenziale e il suo corrispondente set di geni hMSC espressi in modo differenziale. Come tale, è possibile che i geni che sono vitali nella proliferazione del corno di cervo e/o nella mineralizzazione, ma che non sono espressi in modo differenziato, non vengano rilevati. Per affrontare questo, il nostro studio ha adottato una strategia di replicazione biologica limitata (un isolato, due replicati) con un elevato numero di letture di sequenziamento. Questa strategia si basa sull’utilizzo di un gran numero di letture di sequenziamento per generare una maggiore potenza statistica per la rilevazione sensibile dell’espressione genica differenziale[35]. Una strategia di questo tipo sarebbe particolarmente importante per scoprire trascrizioni inedite e con un basso numero di copie. In effetti, questo approccio ha avuto successo nell’identificare un gran numero di trascrizioni non rilevate in precedenza relative agli estrogeni nelle cellule tumorali del seno[36]. In terzo luogo, poiché il genoma del cervo è stato sequenziato solo di recente[37], le letture di sequenziamento sono state mappate sul genoma del Bos taurus, strettamente correlato e ben noto. Anche se tale mappatura incrociata tra specie può portare a una perdita di dati di sequenza e di espressione, nonché a un aumento del bias e dell’errore, queste dimensioni dell’effetto sono riportate come piccole all’interno di una finestra di 100 milioni di anni e mostrano una migliore performance di mappatura rispetto all’assemblaggio del trascrittoma de novo[32]. Nonostante ciò, i nostri set di dati RNA-seq dei cervi contengono una grande percentuale di geni non preannunciati, e questo risultato può essere migliorato applicando la mappatura Blast guidata dall’ortologia.
Conclusione
In conclusione, abbiamo sviluppato un modello in vitro per la comparazione RNA-seq tra cellule RM FD e hMSC per semplificare l’analisi di set di dati trascrittomici e per la prima volta per identificare geni unici pertinenti alla rigenerazione delle corna di cervo. La scoperta di questi geni fa progredire la nostra comprensione della biologia del corno di cervo e offre strategie promettenti per una rapida rigenerazione ossea. Prevediamo che una strategia di confronto simile possa essere applicata a quasi tutti i tessuti per identificare i contributi di geni espressi in modo univoco a un fenomeno di interesse.
Metodi
Materiali e metodi dettagliati sono forniti nel file aggiuntivo 1: Informazioni supplementari.
Daino
I tessuti sono stati raccolti dal Lazy Arrow Camatta Ranch (Santa Margarita, CA) e dal Walking Beam Ranch (Santa Paula, CA) in conformità con le linee guida approvate stabilite dal gruppo di lavoro amministrativo sulla cura degli animali da laboratorio dell’Università di Stanford (APLAC 28057). Il tessuto delle corna è stato identificato[14] e raccolto con la digestione enzimatica.
Coltura cellulare
I fibroblasti mesenchimali del mouse C3H10T1/2 (American Type Culture Collection; ATCC, Manassas, VA) sono stati mantenuti in DMEM, 10% FBS, e 1% P/S e utilizzati entro i primi dieci passaggi dalla data di ricezione. Le cellule FD sono state mantenute in DMEM, 10% FBS e 1% P/S e utilizzate tra i passaggi 2 e 8. Le cellule staminali mesenchimali umane (hMSC; Lonza, Svizzera) sono state mantenute secondo le istruzioni del produttore e utilizzate tra i passaggi 4 e 7. Hoechst colorazione (Anaspec, Fremont, CA) è stato utilizzato per monitorare la contaminazione da micoplasma in colture cellulari.
Proliferazione cellulare
Gli studi sulla proliferazione cellulare sono stati eseguiti in DMEM, 10% FBS, 1% P/S, mezzi di crescita delle cellule staminali mesenchimali (Lonza, Svizzera), e mezzi di crescita delle cellule staminali mesenchimali integrati con 10 ng/mL fattore di crescita dei fibroblasti-2 (FGF-2; Peprotech, Rocky Hill, NJ). Le cellule sono state contate utilizzando un contatore di particelle Beckman Coulter Z2 (Beckman Coulter, Brea, CA), e i tempi di raddoppio delle cellule sono stati calcolati utilizzando R-studio (R Studio, Boston, MA, http://www.rstudio.com). Gli studi del ciclo cellulare sono stati eseguiti utilizzando ioduro di propidio / soluzione di RNAse (Cell Signaling Technology, Danvers, MA) su un citometro a flusso BD Aria II e analizzati utilizzando Flowjo 9.7.5 (Flowjo LLC, Ashland, OR, http://www.flowjo.com).
Differenziazione cellulare
La differenziazione adipogenica e condrogenica sono state eseguite utilizzando rispettivamente StemPro Adipogenic (Gibco, Thermo Fisher Scientific, Waltham, MA) e StemPro Chondrogenic Media (Gibco, Thermo Fisher Scientific, Waltham, MA), secondo le istruzioni del produttore. La differenziazione osteogenica è stata eseguita utilizzando DMEM, 10% FBS, 1% PS, e 100 ng/mL BMP-2 (Infuse Bone Graft, Medtronic, Sunnyvale, CA) o DMEM, 10% FBS, 1% P/S, 50μg/mL di acido ascorbico, 10 mM di β-glicerofosfato, 100 ng/mL di BMP-2 e 100 nM di desametasone (Sigma Aldrich, St. Louis, MO). L’espressione genica osteogenica è stata eseguita su modelli di cDNA (isolamento dell’RNA: Qiagen RNeasy Plus Mini kit, Qiagen, Germania; trascrizione inversa: Omniscript kit, Qiagen, Germania) per 40 cicli utilizzando TaqMan Gene Expression Mastermix (4369016, Applied Biosystems, Thermo Fisher Scientific, Waltham, MA) su un termociclatore Applied Biosystems HT7200 (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA). I dati di espressione genica sono stati analizzati utilizzando SDS 2.2.2 (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com/). L’attività ALP (Kit 86C, Sigma Aldrich, St. Louis, MO) è stata rilevata secondo le istruzioni del produttore. Dove necessario, l’intensità media dei pixel è stata determinata utilizzando lo strumento di istogramma dell’immagine in Adobe Photoshop come descritto in precedenza[38, 39]. La mineralizzazione osteogenica è stata determinata utilizzando il 2% di Alizarin Red S stain (Electron Microscopy Sciences, Hatfield, PA) su campioni estratti da solventi. I valori di assorbanza dei campioni e degli standard sono stati letti a 405 nm utilizzando uno spettrometro Tecan Infinite F50 (Tecan Trading AG, Svizzera).
RNA-seq
Biblioteche di proliferazione sono stati ottenuti da cellule coltivate sotto DMEM, 0% FBS, e 1% P/S (0% siero) e DMEM, 10% FBS, 1% P/S (10% siero) mentre le biblioteche di mineralizzazione sono stati ottenuti da cellule coltivate sotto DMEM, 10% FBS, 1% P/S, 50 μacido ascorbico g/mL, e 10 mM β-glicerofosfato (mezzi di controllo; senza BMP-2 e desametasone) e DMEM, 10% FBS, 1% P/S, 50 μg/mL di acido ascorbico, 10 mM β-glicerofosfato, 100 ng/mL BMP-2, e 100 nM desametasone. L’RNA è stato isolato (kit Qiagen RNeasy Plus Mini, Qiagen, Germania), trascritto al contrario in cDNA (kit Ovation RNA-seq System V2, NuGEN, San Carlos, CA), tosato (S2 Focused-ultrasonicator, Covaris, Woburn, MA), riparato, con coda dA, legato con adattatori personalizzati e amplificato in PCR per la costruzione di biblioteche con RNA-seq (NEBNext DNA Library Prep Master Mix Set per Illumina, New England Biolabs, Ipswich, MA). I campioni sono stati sequenziati utilizzando 100 coppie di basi, tecnologia RNA-seq paired-end (HiSeq 2000, Illumina, San Diego, CA), e i dati sono stati analizzati utilizzando diversi software bioinformatici tra cui Spliced Transcripts Alignment to a Reference (STAR; Versione 2.3.0, https://code.google.com/p/rna-star/)[40], SAMtools (Versione 0.1.19 http://www.htslib. org/), il pacchetto Cufflinks (Versione 2.1.1.1.1, https://github.com/cole-trapnell-lab/cufflinks)[41], il pacchetto Cummerbund (http://compbio.mit.edu/cummeRbund/), Ingenuity Pathway Analysis (Qiagen, Germania; https://www.qiagenbioinformatics.com/), e Gene Ontology Enrichment Analysis (http://geneontology.org/page/go-enrichment-analysis). Le analisi statistiche per l’RNA-seq sono state eseguite come descritto in precedenza[41]. I candidati genetici sono stati identificati sulla base di geni espressi in modo differenziato che hanno mostrato più di cinque volte l’upregulation nel controllo rispetto alle condizioni di trattamento così come i geni che sono stati espressi in modo univoco nel set di dati delle cellule RM FD. Da questo insieme di candidati, i geni del cervo uhrf1 e s100a10 sono stati identificati manualmente come geni di interesse sulla base di una ricerca in letteratura (Pubmed; http://www.pubmed.com) delle loro funzioni biologiche conosciute e novità nel contesto della biologia ossea dei mammiferi.
Clonazione e transfezione dei geni
I geni sono stati clonati mediante PCR (Platinum Blue PCR SuperMix, Invitrogen, Thermo Fisher Scientific, Waltham, MA) dalle biblioteche di cDNA dei cervi utilizzando primer progettati dal genoma bovino strettamente correlato prima della subclonazione in un plasmide pVitro2-MCS-Blast (InvivoGen, San Diego, CA). C3H10T1/2 cellule sono state trasfettate con 2-3 μg di plasmide (s) contenente il genoma di interesse secondo le istruzioni del produttore (Polyplus, Francia), e le cellule staticamente trasfettate sono state selezionate utilizzando 3 μg / ml blasticidina (Invitrogen, Thermo Fisher Scientific, Waltham, MA).
siRNA
Le cellule sono state trasfettate con 30 nM uhrf1 siRNA A ed E (i siRNA sono stati progettati su misura da Santa Cruz Biotechnology Inc., Dallas, TX, sulla base della sequenza uhrf1 bovina) secondo le istruzioni del produttore (Polyplus, Francia) per 72 ore.
Colorazione di immunofluorescenza
Le cellule sono state fissate in paraformaldeide al 4% (Electron Microscopy Sciences, Hatfield, PA), bloccate con siero d’asino al 10% (Jackson Immunoresearch Laboratories Inc., West Grove, PA), e incubate con anticorpo primario seguito da incubazione di anticorpi secondari con opportuni lavaggi nel mezzo. Gli anticorpi comprendevano 10 μg/mL di topo anti-Stro-1 (MAB1038, R&D Systems Inc., Minneapolis, MN), 4 μg/mL di coniglio anti fosfatasi alcalina (Sc-98652, ALP; Santa Cruz Biotechnology Inc., Dallas, TX), 10 μg/mL di coniglio anti-UHRF1 (Sc98704, Santa Cruz Biotechnology Inc, Dallas, TX), 1 μg/mL di topo anti-S100A10 (Ab89438, Abcam Inc., Cambridge, MA), 15 μg/mL di asino anti-topo Alexa 488 (715-545-150, Jackson Immunoresearch Laboratories Inc, West Grove, PA), e 15 μg/mL asino anti-coniglio Alexa 647 (711-605-152, Jackson Immunoresearch Laboratories Inc., West Grove, PA). Dove necessario, il recupero dell’antigene è stato eseguito utilizzando la soluzione tampone di recupero dell’antigene (IHC World LLC, Woodstock, MD) a 80-90 °C per 30-60 minuti prima dell’incubazione dell’anticorpo. L’intensità media dei pixel è stata determinata utilizzando lo strumento di istogramma dell’immagine in Adobe Photoshop come descritto in precedenza[38, 39].
Analisi statistica
Le analisi statistiche che coinvolgono RNA-seq sono state effettuate da Cufflinks e R-Studio[41]. La significatività statistica per i geni espressi in modo differenziato è stata stabilita a p≤ 0,05 e q≤ 0 ,05. Le analisi statistiche che non coinvolgono RNA-seq sono state effettuate utilizzando le IBM SPSS Statistics for Windows 22 (IBM Corp., North Castle, NY, http://www.ibm.com). I dati quantitativi sono stati presentati come errore di media (mean±standard error of mean (mean±SEM) ove opportuno. I cambiamenti relativi di piega per i dati di PCR sono stati trasformati in log per rendere la distribuzione dei dati più simmetrica[42]. Il test di Shapiro-Wilk e il test di Levene sono stati utilizzati per determinare se i dati erano normalmente distribuiti e contenevano variazioni uguali tra i gruppi, rispettivamente. Per due confronti medi, i valori p sono stati calcolati tramite il test t. I valori p sono stati calcolati utilizzando la varianza raggruppata e separata per i dati con varianze uguali e disuguali, rispettivamente. Per più di due confronti medi, i valori p sono stati calcolati tramite l’analisi della varianza (ANOVA). Se la maggior parte dei dati era normalmente distribuita o c’era una varianza uguale tra i gruppi, i valori di p sono stati calcolati usando ANOVA seguito dal test di confronto multiplo onesto della differenza significativa post hoc di Tukey [43,44]. Altrimenti, i valori p sono stati calcolati usando l’ANOVA di Welch seguito dal test di confronto multiplo post hoc di Games-Howell [45]. La significatività statistica è stata stabilita a p≤ 0,05.
File aggiuntivi
File aggiuntivo 1:Cifre supplementari. (PDF 2760 kb)File aggiuntivo 2:Confronto tra i geni espressi in modo diverso dal Daino RM e le cellule staminali mesenchimali umane. (XLSX 13100 kb)
References
- Gómez JA, Ceacero F, Landete-Castillejos T, Gaspar-López E, García AJ, Gallego L. Factors affecting antler investment in Iberian red deer. Anim Prod Sci. 2012; 52:867-873. DOI
- Huxley JS. The relative size of antlers in deer. Proc Zool Soc London. 1931; 101:819-864. DOI
- Goss RJ. Deer antlers. Regeneration, function and evolution. Academic Press: New York; 1983.
- Kierdorf U, Kierdorf H. Deer antlers – a model of mammalian appendage regeneration: an extensive review. Gerontology. 2011; 57:53-65. DOI | PubMed
- Li C. Histogenetic aspects of deer antler development. Front Biosci. 2013; 5:479-489. DOI
- Li C, Yang F, Sheppard A. Adult stem cells and mammalian epimorphic regeneration-insights from studying annual renewal of deer antlers. Curr Stem Cell Res Ther. 2009; 4:237-251. DOI | PubMed
- Price J, Faucheux C, Allen S. Deer antlers as a model of mammalian regeneration. Curr Top Dev Biol. 2005; 67:1-48. DOI | PubMed
- Sissons HA, Kember NF. Longitudinal bone growth of the human femur. Postgrad Med J. 1977; 53:433-437. DOI | PubMed
- Allen SP, Maden M, Price JS. A role for retinoic acid in regulating the regeneration of deer antlers. Dev Biol. 2002; 251:409-423. DOI | PubMed
- Bartoš L, Schams D, Bubenik GA. Testosterone, but not IGF-1, LH, prolactin or cortisol, may serve as antler-stimulating hormone in red deer stags (Cervus Elaphus). Bone. 2009; 44:691-698. DOI | PubMed
- Daley E, Alford A, Miller JD, Goldstein S. Phenotypic differences in white-tailed deer antlerogenic progenitor cells and marrow-derived MSC. Tissue Eng A. 2013; 20:1416-1425. DOI
- Gyurján I, Molnár A, Borsy A, Stéger V, Hackler L, Zomborszky Z, Papp P, Duda E, Deák F, Lakatos P. Gene expression dynamics in deer antler: mesenchymal differentiation toward chondrogenesis. Mol Gen Genomics. 2007; 277:221-235. DOI
- Hu W, Li T, Hu R, Wu L, Li M, Meng X. MicroRNA let-7a and let-7f as novel regulatory factors of the sika deer (Cervus Nippon) IGF-1R gene. Growth Factors. 2014; 32:27-33. DOI | PubMed
- Li C, Clark DE, Lord EA, Stanton JA, Suttie JM. Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery. Anat Rec. 2002; 268:125-130. DOI | PubMed
- Molnár A, Gyurján I, Korpos É, Borsy A, Stéger V, Buzás Z, Kiss I, Zomborszky Z, Papp P, Deák F, Orosz L. Identification of differentially expressed genes in the developing antler of red deer Cervus elaphus. Mol Gen Genomics. 2007; 277:237-248. DOI
- Price JS, Oyajobi BO, Oreffo RO, Russell RG. Cells cultured from the growing tip of red deer antler express alkaline phosphatase and proliferate in response to insulin-like growth factor-I. J Endocrinol. 1994; 143:R9-16. DOI | PubMed
- Rolf HJ, Kierdorf U, Kierdorf H, Schulz J, Seymour N, Schliephake H, Napp J, Niebert S, Wolfel H, Wiese KG. Localization and characterization of STRO-1 cells in the deer pedicle and regenerating antler. PLoS One. 2008; 3:e2064. DOI | PubMed
- Yao B, Zhao Y, Wang Q, Zhang M, Liu M, Liu H, Li J. De novo characterization of the antler tip of Chinese sika deer transcriptome and analysis of gene expression related to rapid growth. Mol Cell Biochem. 2012; 364:93-100. DOI | PubMed
- Yao B, Zhao Y, Zhang H, Zhang M, Liu M, Liu H, Li J. Sequencing and de novo analysis of the Chinese sika deer antler-tip transcriptome during the ossification stage using Illumina Rna-Seq technology. Biotechnol Lett. 2012; 34:813-822. DOI | PubMed
- Zhao Y, Yao B, Zhang M, Wang S, Zhang H, Xiao W. Comparative analysis of differentially expressed genes in sika deer antler at different stages. Mol Biol Rep. 2012; 40:1665-1676. DOI | PubMed
- Bae KM, Kwon YS, Cho IH, Yi SI. Use of cDNA-AFLP for transcript profiling in narrow genetic pools; for example, cucumber (Cucumis sativus L.). Plant Breed. 2006; 125:488-492. DOI
- Mendicino M, Bailey AM, Wonnacott K, Puri RK, Bauer SR. MSC-based product characterization for clinical trials: an FDA perspective. Cell Stem Cell. 2014; 14:141-145. DOI | PubMed
- Bartmann C, Rohde E, Schallmoser K, Purstner P, Lanzer G, Linkesch W, Strunk D. Two steps to functional mesenchymal stromal cells for clinical application. Transfusion. 2007; 47:1426-1435. DOI | PubMed
- Schallmoser K, Bartmann C, Rohde E, Reinisch A, Kashofer K, Stadelmeyer E, Drexler C, Lanzer G, Linkesch W, Strunk D. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion. 2007; 47:1436-1446. DOI | PubMed
- Ker DFE, Sharma R, Wang ETH, Yang YP. Development of mRuby2-transfected C3H10T1/2 fibroblasts for musculoskeletal tissue engineering. PLoS One. 2015; 10:e0139054. DOI | PubMed
- Sharif J, Muto M, S-i T, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K. The SRA protein NP95 mediates epigenetic inheritance by recruiting DNMT1 to methylated DNA. Nature. 2007; 450:908-912. DOI | PubMed
- Mousli M, Hopfner R, Abbady AQ, Monte D, Jeanblanc M, Oudet P, Louis B, Bronner C. ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br J Cancer. 2003; 89:120-127. DOI | PubMed
- Zhao J, Chen X, Song G, Zhang J, Liu H, Liu X. UHRF1 controls the self-renewal versus differentiation of hematopoietic stem cells by epigenetically regulating the cell-division modes. Proc Natl Acad Sci. 2017; 114:E142-E151. DOI | PubMed
- Hajjar KA. The biology of Annexin A2: from vascular fibrinolysis to innate immunity. Trans Am Clin Climatol Assoc. 2015; 126:144-155. PubMed
- Duarte WR, Shibata T, Takenaga K, Takahashi E, Kubota K, Ohya K, Ishikawa I, Yamauchi M, Kasugai S. S100A4: a novel negative regulator of mineralization and osteoblast differentiation. J Bone Miner Res. 2003; 18:493-501. DOI | PubMed
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008; 5:621-628. DOI | PubMed
- Hornett EA, Wheat CW. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC Genomics. 2012; 13:361. DOI | PubMed
- Diederichs S, Tuan RS. Functional comparison of human-induced pluripotent stem cell-derived mesenchymal cells and bone marrow-derived mesenchymal stromal cells from the same donor. Stem Cells Dev. 2014; 23:1594-1610. DOI | PubMed
- Obata Y, Furusawa Y, Endo TA, Sharif J, Takahashi D, Atarashi K, Nakayama M, Onawa S, Fujimura Y, Takahashi M. The epigenetic regulator UHRF1 facilitates the proliferation and maturation of colonic regulatory T cells. Nat Immunol. 2014; 15:571-579. DOI | PubMed
- Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-Seq: a matter of depth. Genome Res. 2011; 21:2213-2223. DOI | PubMed
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011; 145:622-634. DOI | PubMed
- Bana NA, Nyiri A, Nagy J, Frank K, Nagy T, Steger V, Schiller M, Lakatos P, Sugar L, Horn P. The red deer Cervus Elaphus genome Cerela1.0: sequencing, annotating, genes, and chromosomes. Mol Gen Genomics. 2018; 293:665-684. DOI
- Ker ED, Chu B, Phillippi JA, Gharaibeh B, Huard J, Weiss LE, Campbell PG. Engineering spatial control of multiple differentiation fates within a stem cell population. Biomaterials. 2011; 32:3413-3422. DOI | PubMed
- Ker ED, Nain AS, Weiss LE, Wang J, Suhan J, Amon CH, Campbell PG. Bioprinting of growth factors onto aligned sub-micron fibrous scaffolds for simultaneous control of cell differentiation and alignment. Biomaterials. 2011; 32:8097-8107. DOI | PubMed
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal Rna-Seq aligner. Bioinformatics. 2012; 29:15-21. DOI | PubMed
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and Transcript expression analysis of RNA-Seq experiments with Tophat and cufflinks. Nat Protoc. 2012; 7:562-578. DOI | PubMed
- Derveaux S, Vandesompele J, Hellemans J. How to do successful gene expression analysis using real-time PCR. Methods. 2010; 50:227-230. DOI | PubMed
- Box GEP. Non-normality and tests on variances. Biometrika. 1953; 40:318-335. DOI
- Box GEP. Some theorems on quadratic forms applied in the study of analysis of variance problems, I. effect of inequality of variance in the one-way classification. Ann Math Stat. 1954; 25:290-302. DOI
- Tomarken Andrew J., Serlin Ronald C.. Comparison of ANOVA alternatives under variance heterogeneity and specific noncentrality structures. Psychological Bulletin. 1986; 99(1):90-99. DOI
Fonte
Ker DFE, Wang D, Sharma R, Zhang B, Passarelli B, et al. (2018) Identifying deer antler . Stem Cell Research & Therapy 9292. https://doi.org/10.1186/s13287-018-1027-6




